ISO 14937, titled “Sterilization of Health Care Products — General Requirements for Characterization of a Sterilizing Agent and the Development, Validation and Routine Control of a Sterilization Process for Medical Devices,” outlines essential guidelines for ensuring the efficacy and safety of sterilization processes used in healthcare product manufacturing. This standard provides comprehensive requirements for characterizing sterilizing agents and establishing, validating, and maintaining sterilization processes for medical devices. By adhering to ISO 14937, manufacturers can ensure the consistent and reliable sterilization of medical devices, thereby minimizing the risk of infection and promoting patient safety. This standard serves as a vital tool for regulatory compliance and quality management within the healthcare industry, facilitating the development and implementation of robust sterilization processes that meet international standards and regulatory requirements.
This article is part of the ongoing work focused on the analysis of ISO standards related to sterilization topics; several of these topics have already been discussed within 4EasyReg, such as gamma sterilization, requirements for biological indicators, ethylenoxide sterilization and much more. In this article, we will deal with a general overview of the requirements related to ISO 14937. We will not be able to provide an in-depth overview of the full standard, this we are going to select only specific aspects which we are going to discuss in the next sections of this article.
We have been already discussing about different methods for sterilization, such as ethylene oxide, gamma sterilization, and related topics such as biological indicators, packaging requirements for sterile devices and much more.

Quality management System Requirements according to ISO 14937
An overview of the QMS related requirements in relation to ISO 14937 is reported in the scheme below.
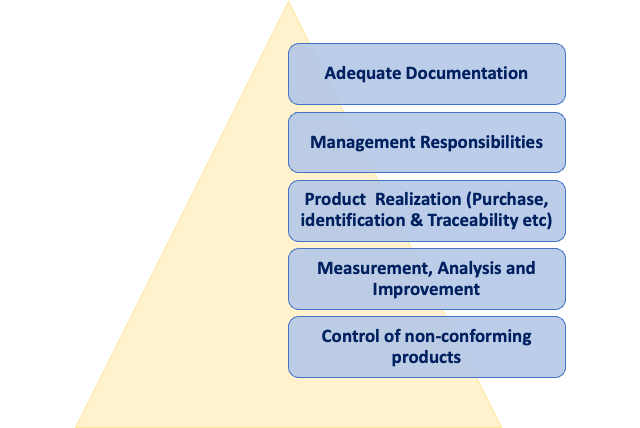
Procedures for the characterization of a sterilizing agent, development, validation, and routine control of a sterilization process, as well as product release from sterilization, shall be drafted; as usual, all the records related to the aforementioned procedures shall undergo review and approval by designated personnel and be controlled in accordance with the applicable clauses of ISO 13485.
The responsibility and authority for implementing and meeting the requirements of ISO 14937 must be clearly specified, with competent personnel assigned responsibility in alignment with the relevant clauses of ISO 13485. If organizations with separate quality management systems undertake the requirements of ISO 14937, the responsibilities and authority of each party must be specified.
Furthermore, it is essential that procedures for purchasing, product identification, and traceability must be specified to comply with the applicable clauses of ISO 13485. A calibration system for all equipment, including test instrumentation, used in meeting the requirements of this International Standard, must be specified to comply with the applicable clause(s) of ISO 13485 or ISO 10012. Procedures for the control of non-conforming product, along with correction, corrective action, and preventive action, must be specified, complying with the applicable clauses of ISO 13485.
-
 Process Validation Procedure€64,00
Process Validation Procedure€64,00
Requirements for the characterisation of the sterilising agents
The primary objective of this phase is to delineate the sterilizing agent, assess its efficacy against microorganisms, evaluate its impact on materials, and ensure safety for personnel and the environment. These activities may involve testing in a prototype system, with subsequent alignment of equipment specifications with experimental findings.
The sterilizing agent must be precisely specified, including conditions for storage to maintain its effectiveness over its shelf life. Microbicidal effectiveness studies must encompass several facets, including demonstrating lethal action against representative microorganisms, establishing mathematical models for microbial inactivation kinetics, identifying resistant microorganisms, and assessing factors that may influence effectiveness and delivery of the sterilizing agent. Test methods, acceptance criteria, and choice of test microorganisms must be documented and recorded. Furthermore, the effects of sterilizing agent exposure on material properties, including biological safety, must be thoroughly evaluated, along with considerations for repeated exposure.
Safety and environmental concerns necessitate the specification of safety information for the sterilizing agent and its precursors, ensuring compliance with regulatory standards and facilitating experimental studies.
Routing monitoring & control of the sterilization process as per ISO 14937
Routine monitoring and control serve the purpose of verifying that the validated sterilization process has effectively treated the product. This entails obtaining evidence, primarily through measurements, supplemented by biological or chemical indicators if necessary, to ensure that the sterilization process adhered to defined tolerances. Recorded data must demonstrate the attainment of process parameters within these predefined tolerances: obviously, all relevant records must be diligently retained in accordance with specified requirements.
Biological indicators and chemical indicators, if used, shall comply with the specific requirements mentioned within ISO 14937; additionally, Process Challenge Devices (PCDs), if utilized in routine monitoring and control, must also adhere to the specifications detailed in section 8.6 of ISO 14937. These measures collectively ensure the ongoing effectiveness and consistency of the sterilization process.
Product Release
A defined procedure for releasing products from sterilization must be established and implemented, as specifically requested by Iso 13485; the procedure shall outline the criteria for determining if a sterilization process meets its specifications. Parametric release is permissible only when all process parameters are meticulously specified, controlled, and directly monitored, with detailed records of these parameters retained. Results from biological or chemical indicators, if utilized in monitoring the sterilization process, must also factor into the criteria for product release. Failure to meet the specified criteria renders the product non-conforming, requiring handling in accordance with documented procedures of the quality system of the organization.
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Regulatory matters within the medical device, information security and AI-Based business.
We offer a wide range of documentation kits to support your compliance efforts towards a wide range of standards and regulations, such as ISO 13485, EU MDR, ISO 27001, ISO 42001 and much more. . Specifically, in our webshop you will find:
- ISO 13485 Documentation / Compliance Kit
- EU MDR Documentation Kit
- MDSAP Documentation Kit
- ISO 27001 Documentation / Compliance Kit
- ISO 42001 Documentation / Compliance Kit
- FDA Cybersecurity Documentation
Within our sister platform QualityMedDev Academy, a wide range of online & self-paced training courses is available, such as for example:
- Complaint Handling and Vigilance Reporting
- Artificial Intelligence in Medical Device. Regulatory Requirements
- Unique Device Identification (UDI) Requirements according to EU MDR
- Clinical Evaluation Process According to EU MDR
- Medical Device SW Verification & Validation
- Risk Management for Medical Devices
- Usability Evaluation for Medical Devices
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
