The Food and Drug Administration (FDA) plays a pivotal role in safeguarding public health through the regulation of medical devices, ensuring that they are both safe and effective for their intended use. In contemporary times, the rise of connected devices has brought cybersecurity to the forefront of FDA concerns; in this context, defining specific requirements for the definition of cybersecurity threat model is essential. As medical devices become increasingly integrated with technology, the need for robust cybersecurity measures to protect patient information and ensure the functionality of these devices is more critical than ever. Compliance with FDA regulations is not only essential for the conformity of manufacturers but also for the safety and trust of patients and users.
We have been discussing about digital medical devices and related requirements several times within QualityMedDev websites, covering different topics such as AI within medical devices, digital health products, and TGA approach for the regulation of digital medical devices. At the same time, FDA has published different guidelines in relation to cybersecurity, either in relation to pre-market requirements and post-market requirements.
The FDA has established cybersecurity requirements as part of their regulatory oversight to protect patients and ensure the integrity of medical devices. These standards apply to the full lifecycle of a device, from initial design and development to deployment and maintenance. With medical devices becoming smarter and more connected, they are increasingly vulnerable to cyber threats. The integration of cybersecurity practices during the development phase of medical devices is a crucial step for mitigating risks posed by cyber attacks.
Key Components of the FDA’s Cybersecurity Framework
To ensure the cybersecurity integrity of medical devices, the FDA has outlined pre-market requirements that include the need for manufacturers to incorporate cybersecurity threat model and security controls from the earliest stages of device conception.
Manufacturers of devices shall establish and adhere to quality systems to ensure consistent compliance with applicable requirements and specifications for their products. The requirements for these quality systems can be found in the Quality System (QS) regulation in 21 CFR Part 820 if the device is sold in United States, or other regulations for other countries (e.g. EU MDR 2017/745 for European Union).
Depending on the nature of the device, the QS requirements may be pertinent during the premarket stage, postmarket stage, or both. In the context of the premarket phase, demonstrating reasonable assurance of safety and effectiveness for certain devices with cybersecurity risks may involve including documentation outputs related to the QS regulation as part of the premarket submission. For instance, ISO 13485:2016 and 21 CFR 820 mandates that manufacturers of all classes of devices automated with software establish procedures to control the design of the device, ensuring compliance with specified design requirements (referred to as “design controls”). Within design controls, manufacturers are required to “establish and maintain procedures for validating the device design,” which encompasses “software validation and risk analysis, where appropriate” . As part of the mandated software validation and risk analysis, software device manufacturers may need to institute cybersecurity risk management and validation processes when applicable.
Software validation and risk management constitute crucial elements of cybersecurity analyses, determining whether a device provides reasonable assurance of safety and effectiveness. The FDA mandates manufacturers to incorporate development processes that consider and address software risks throughout the design and development stages as part of design controls. These processes should encompass cybersecurity considerations. This includes addressing the identification of security risks, establishing design requirements for controlling these risks, and providing evidence that the controls operate as intended and are effective in the device’s designated environment, ensuring sufficient security measures.
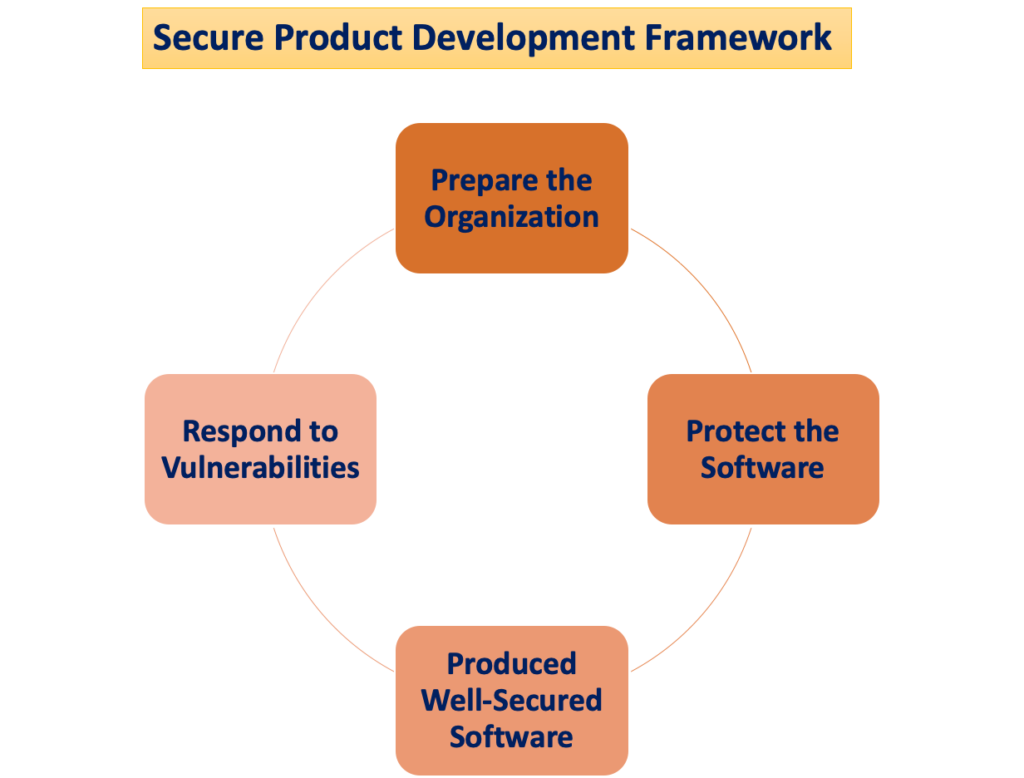
The “Secure Product Development Framework“
The potential for patient harm arises when cybersecurity threats exploit vulnerabilities in a system, and the ease with which these threats can compromise the safety and effectiveness of a medical device increases with the number of identified vulnerabilities over time. A Secure Product Development Framework (SPDF) consists of processes aimed at identifying and diminishing the quantity and severity of vulnerabilities in products. Encompassing all stages of a product’s lifecycle—design, development, release, support, and decommissioning—the SPDF is integral.
During device design, incorporating SPDF processes can prevent the necessity for re-engineering when integrating post-marketing connectivity-based features or addressing vulnerabilities that pose uncontrolled risks. Feasible integration with existing processes for product and software development, risk management, and the broader quality system adds to the versatility of the SPDF.
To ensure compliance with the Quality System (QS) regulation, the use of an SPDF is recommended. The FDA encourages manufacturers to embrace the SPDF for its benefits in meeting both the QS regulation and cybersecurity requirements. It is acknowledged, however, that alternative approaches may also satisfy the QS regulation.
Management of Cybersecurity Risks
The primary objective of employing an SPDF (Secure Product Development Framework) is to create and sustain devices that are both safe and effective. From a security perspective, these devices also earn trustworthiness and resilience. Manufacturers and/or users (e.g., patients, healthcare facilities) can then manage these devices, including installation, configuration, updates, and review of device logs, through the device design and associated labeling.
Healthcare facilities have the option to manage these devices within their own cybersecurity risk management frameworks, such as the widely recognized National Institute of Standards and Technology (NIST) Framework for Improving Critical Infrastructure Cybersecurity, commonly referred to as the NIST Cybersecurity Framework or NIST CSF.
The FDA recommends that manufacturers incorporate device design processes, such as those outlined in the Quality System (QS) regulation, to bolster secure product development and maintenance. While maintaining flexibility for manufacturers, they may also explore alternative frameworks that align with the QS regulation and adhere to FDA recommendations for implementing an SPDF. Examples include the medical device-specific framework in the Medical Device and Health IT Joint Security Plan (JSP) 30 and IEC 81001-5-1. Frameworks from other sectors, such as the ANSI/ISA 62443-4-1 Security for industrial automation and control systems Part 4-1: Product security development life-cycle requirements, may also meet QS regulations.
In the following sections of this article, recommendations for utilizing SPDF processes are provided, as generally perceived by the FDA, which highlight crucial considerations for developing devices that are safe and effective. These processes complement the QS regulation, and the FDA suggests manufacturers include corresponding documentation for review in premarket submissions.
Cybersecurity Threat Model
Threat modeling involves a systematic process for identifying security objectives, risks, and vulnerabilities throughout the medical device system. Subsequently, it entails defining countermeasures to prevent, mitigate, monitor, or respond to the impacts of threats throughout the lifecycle of the medical device system. When appropriately and comprehensively applied, it serves as the foundation for optimizing security across system components, products, networks, applications, and connections.
Regarding security risk management, and to pinpoint suitable security risks and controls for the medical device system, the FDA advocates for the implementation of threat modeling to inform and support risk analysis activities. In the context of risk assessment, the FDA suggests integrating threat modeling throughout the design process, encompassing all elements of the medical device system.
Key aspects of the threat model should encompass the identification of risks and mitigations, informing both pre- and post-mitigation risks considered in the cybersecurity risk assessment. Additionally, the model should articulate assumptions about the medical device system or the usage environment, such as assuming hospital networks are inherently hostile. This assumption prompts manufacturers to consider scenarios where an adversary controls the network, capable of altering, dropping, and replaying packets. Furthermore, the threat model should capture cybersecurity risks introduced through the supply chain, manufacturing, deployment, interoperation with other devices, maintenance/update activities, and decommission activities, which might be overlooked in a traditional safety risk assessment process.
For premarket submissions, the FDA recommends including threat modeling documentation to showcase the analysis of the medical device system, identifying potential security risks that could impact safety and effectiveness. Manufacturers have the flexibility to choose from various methodologies or combinations of methods for threat modeling, and the rationale for their chosen methodologies should be provided with the documentation.
Conducting threat modeling activities during design reviews is advised, and the documentation should furnish sufficient information for the FDA to assess and review the security features integrated into the device. This holistic evaluation should consider both the device and the broader system in which it operates, emphasizing the safety and effectiveness of the device.
The main elements of the cybersecurity threat models can be summarised as follows:
Identifying Potential Threats
The development of a threat model serves as a foundational step in the cybersecurity process, enabling manufacturers to identify and understand potential cybersecurity threats specific to their medical devices. The development of a threat model serves as a foundational step in the cybersecurity process, enabling manufacturers to identify and understand potential cybersecurity threats specific to their medical devices. The development of a threat model serves as a foundational step in the cybersecurity process, enabling manufacturers to identify and understand potential cybersecurity threats specific to their medical devices.
Risk Assessment and Management
Risk assessment and management involve evaluating the identified threats to establish their potential impact and devising appropriate strategies to mitigate these risks effectively.
Implementation of Security Controls
Security controls are the safeguards or countermeasures implemented to protect the confidentiality, integrity, and availability of the medical device from identified threats.
Future Trends in FDA Cybersecurity Guidelines
As threats and technology evolve, so too will the FDA’s cybersecurity guidelines. It is essential for manufacturers to remain informed and agile in the face of these changes. Manufacturers should anticipate updates to regulations by staying abreast of industry trends and maintaining a proactive approach to cybersecurity.
Preparation for unforeseen challenges is key; establishing robust security protocols and forward-looking strategies will position manufacturers to effectively respond to the future state of cybersecurity regulations.
Cybersecurity is an aspect of medical device regulation that cannot be overstated—it is as intrinsic to device safety as any mechanical or electrical feature. The FDA has recognized this and by implementing a comprehensive cybersecurity framework, it aims to keep pace with innovation while protecting public health. Manufacturers, healthcare professionals, and patients must all collaborate to uphold these standards and ensure the continued reliability and safety of medical devices in the face of cyber threats.
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Quality & Regulatory matters within the medical device industry. Have a look to all the services that we provide: we are very transparent in the pricing associated to these consulting services.
Within our WebShop, a wide range of procedures, templates, checklists are available, all of them focused on regulatory topics for medical device compliance to applicable regulations. Within the webshop, a dedicated section related to cybersecurity and compliance to ISO 27001 for medical device organizations is also present.
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
