The risk management plan is one of the key document for the risk management process for a medical device manufacturers and it is essential to understand the requirements that are necessary to cover according to ISO 14971:2019 and ISO/TR 24971:2020. Within 4EasyReg websites, we have been discussing several topics related to risk management, including risk management requirements for other life science sectors, for example the pharmaceutical sector and the related ICH Q9 guideline.
An Introduction to the Risk Management Plan
The risk management plan is a document that defines the activities, responsibilities and criteria for risk acceptability for the risk management process. It is usually part of the technical documentation of a medical device.
It is a document that needs to be updated during the whole lifecycle of the device and it interacts with different other processes of the quality system of the organization such as clinical evaluation, post-market surveillance, vigilance reporting, and of course design and development.
-
 Risk Management Report Template€64,00
Risk Management Report Template€64,00 -
 Risk Management Plan Template€64,00
Risk Management Plan Template€64,00
What is the scope of the Risk Management Plan
It is essential to document the life cycle of the medical device along with the risk management activities to be performed.
An example of product life cycle with the related phase of the risk management process can be the one defined below:
| Product Life Cycle | Risk Management Activities |
| Design | Risk Analysis, Risk Evaluation, Risk Control and Residual Risk Acceptability |
| Manufacturing, Logistic and Shelf Life | Production and post-production risk management |
| Use and device end of life | Production and post-production risk management |
Responsibilities and authorities
Responsibilities for the execution of specific risk management activities shall be defined in the risk management plan. Furthermore, it is essential to identify responsibilities for the review and approval of risk management decisions. In case of medical devices containing software, specific competencies both in software development and risk management are needed.
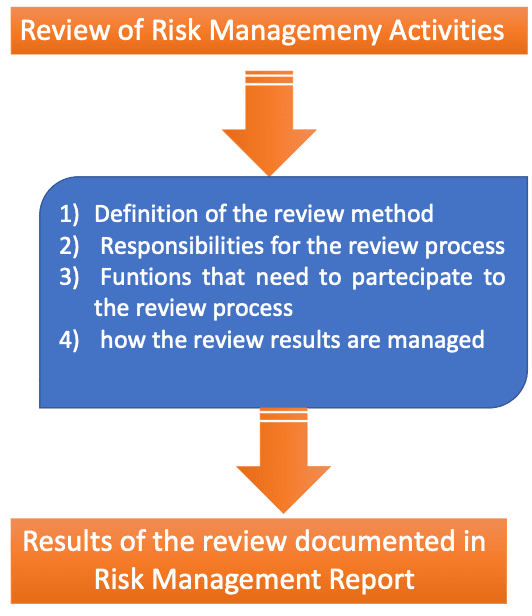
Review of Risk Management Activities
The risk management plan shall include specific methodologies for the review of risk management activities. In the scheme below, more details on the review process are highlighted.
Risk Acceptability
Risks acceptability criteria are of central importance in the risk management plan. The medical device manufacturer shall define the criteria for risk acceptability based on a specific policy for risk acceptability. It is up to the organization to device whether the same risk acceptability policy shall be applied to a specific medical device or the same criteria can be applied to all medical devices.
For software-based medical devices, a different risk acceptability criteria might be needed, because the probability of the harm cannot be estimated. In this case the risk acceptance criteria should be based on the severity of the harm.
In these situations where where probability cannot be estimated, the risk acceptance criteria for residual risk, should take into account the risk control measures that have been implemented and the effectiveness of those risk control measures in reducing the probability of occurrence of harm.
Verification of Risk Control Measures
Verification of risk control measures shall also be defined in the risk management plan for medical devices according to ISO 14971.
According to ISO 14971:2019, two types of verification activities need to be performed:
- Verification of implementation of risk control measures
- Verification of the effectiveness of risk control measures
Both these verification activities can be performed in different ways, for example through design review, design specifications or design and development verification in a quality management system.
-
 Risk Management Package€149,00
Risk Management Package€149,00
Production and Post-Production Risk Management
It is of fundamental importance to establish a solid process for the collection of production and post-production information that can be used to feed the risk management process. The amount of information could be substantially big thus the organization shall have a soldi process in place to handle the analysis of this information and to actively identify trends. Statistical techniques should be considered to assist in the processing of the collected data.
Post-market surveillance plays of course the main role for the collection of information in the post-production phase, and the ISO 20416 could be extremely helpful in the organisation of an efficient post-market surveillance process. Sometimes, for some medical devices, it is essential to conduct post-market clinical follow-up studies, the result of which can be used to identify novel or unidentified risks.

4EasyReg Risk Management Documentation
Nobody can deny the importance of risk management in the medical device field. The regulation in the last 10 years shifted completely towards a situation where risk management process is at the core of quality management system and technical documentation for medical devices. In order to support the implementation of an efficient risk management process, 4EasyReg provides different documentation which can definitely help your organization in the implementation, reorganisation or improvement of risk management. In fact, in the 4EasyReg DocShop, the following documentation can be downloaded:
- Risk Management Procedure , to ensure that the risk management process is well defined within your Quality Management System
- Risk Management Plan Template , which can be used as starting point for the practical implementation of the risk management process
- Risk Analysis Template, which provides you a great example of template that can be used for your risk analysis.
Conclusions
In conclusions, the risk management plan is one of the essential document for the risk management plan. In this post we have been discussing in details what should be addressed within the risk management plan and the interactions of this process with other quality management system processes such as clinical validation and post-market surveillance.
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Regulatory matters within the medical device, information security and AI-Based business.
We offer a wide range of documentation kits to support your compliance efforts towards a wide range of standards and regulations, such as ISO 13485, EU MDR, ISO 27001, ISO 42001 and much more. . Specifically, in our webshop you will find:
- ISO 13485 Documentation / Compliance Kit
- EU MDR Documentation Kit
- MDSAP Documentation Kit
- ISO 27001 Documentation / Compliance Kit
- ISO 42001 Documentation / Compliance Kit
- FDA Cybersecurity Documentation
Within our sister platform QualityMedDev Academy, a wide range of online & self-paced training courses is available, such as for example:
- Complaint Handling and Vigilance Reporting
- Artificial Intelligence in Medical Device. Regulatory Requirements
- Unique Device Identification (UDI) Requirements according to EU MDR
- Clinical Evaluation Process According to EU MDR
- Medical Device SW Verification & Validation
- Risk Management for Medical Devices
- Usability Evaluation for Medical Devices
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
