SAMD – Software As Medical Device became more and more spread within the healthcare. The worldwide spread of software applications pushed the regulators to include in medical device regulations specific requirements for SAMD.
Software is somehow complex and it is a topic we have already quite extensively treated at 4Easyreg. We have shown, for example, of SW related technical documentation, like the Software Development Plan. Or we have been talking about the strategy for software validation, like GAMP-5 approach, with an e-book that can be easily downloaded from our shop.
Here in this post we will go through the concept of SAMD (Software as medical device) with a specific emphasis on the EU MDR regulation.
Let’s start with some definitions. According to the IMDRF – International Medical Device Regulators Forum, the term “Software as a Medical Device” (SaMD) is defined as software intended to be used for one or more medical purposes that perform these purposes without being part of a hardware medical device. In this article we will go through the main characteristics and requirements associated to Software As Medical Devices.
Similarly to classic medical devices, SaMD are subject to all the standard regulations for the medical device sector, including risk management, usability and product verification and validation.
Some Definitions
We are going to start with some definitions in order to fully understand what is the approach used for the regulation of SAMD according to EU MDR 2017/745 and EU IVDR 2017/746.
- Software : a set of instructions that processes input data and creates output data.
- Medical Device Software (MDSW) : edical device software is software that is intended to be used, alone or in combination, for a purpose as specified in the definition of a “medical device” in the medical devices regulation or in vitro diagnostic medical devices regulation.
- Medical Device means any instrument, apparatus, appliance, software, implant, reagent, material or other article intended by the manufacturer to be used, alone or in combination, for human beings for one or more of the following specific medical purposes:
- diagnosis, prevention, monitoring, prediction, prognosis, treatment or alleviation of disease,
- diagnosis, monitoring, treatment, alleviation of, or compensation for, an injury or disability,
- investigation, replacement or modification of the anatomy or of a physiological orpathological process or state,
- providing information by means of in vitro examination of specimens derived from the humanbody, including organ, blood and tissue donations,and which does not achieve its principal intended action by pharmacological, immunological or metabolic means, in or on the human body, but which may be assisted in its function by such means.
Is Your Software a Medical Device ?
The first step to be taken in consideration is to establish whether the software can be considered a medical device. Let’s for the moment considered the EU MDR 2017/745. To decide if a software can be considered a medical device the MDCG guidance 2019-11 shall be followed. Specifically, in order to be considered as medical device software, the specific product must fulfil:
- the definition of software
- The definition of medical devices
There is also the possibility that a software does not meet the definition of a medical device but it can be considered, for example, an accessory. In this case, the product perfectly falls under the EU MDR or IVDR regulation.
There are software that are not intended for medical purposes (for example software which is used to support quality management system activities); in this vase the SW does not qualify as a medical device software.
The MDCG guidance 2019-11 provides a specific workflow to decide whether the specific software can be covered by the medical device regulation EU MDR 2017/745 or not.
Medical Device Software (MDSW)
Medical device software (MDSW) is a software which can be used, alone or in combination, for a purpose as specified in the definition of medical device that we have been reported above. There is the possibility that the software alone is considered a medical or that the software is driving a medical device hardware; in the latter case, if the SW as a medical purpose, then it is also consider a medical device.
Categorisation of SaMD
One of the key aspect is the categorisation of SaMD. There are specific principles which are used to determine the categories of the SaMD. Going more in details, a SaMD can be classified in four different categories which are based on the impact on patient or public health where detailed and precise information provided by the Software as a Medical Device to treat or diagnose, drive or inform clinical management is vital to avoid death, long-term disability or other serious deterioration of state of health. Level IV of SaMD has the highest impact on patient or public health whereas Level I is the lowest.
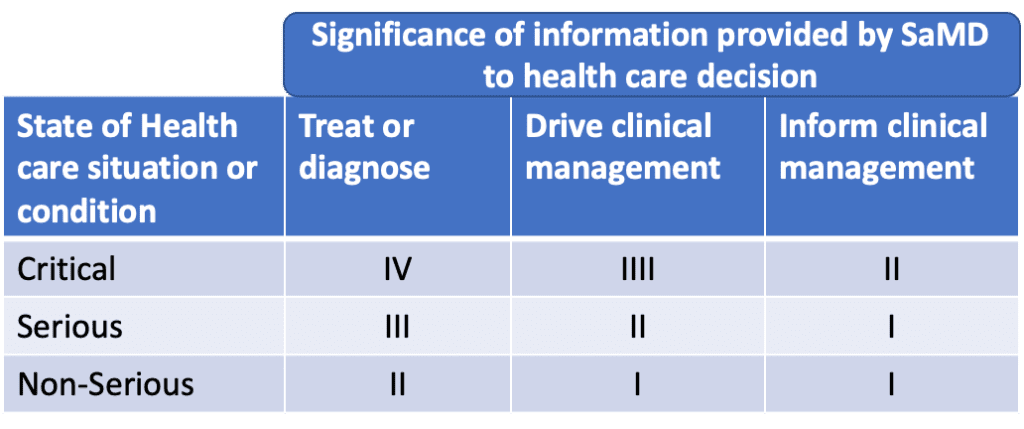
A closer look to SaMD categories
The SaMD of Category IV, as mentioned in the table above, are the ones the provide information to diagnosis or treat critical situations or conditions. Category III are the SaMD similar to category IV. In this case, the software is providing information to support the diagnosis or treatment or serious conditions. Like category IV, the impact of this type of software on patient safety or on public health is classified as high. SaMD that provides information to treat or diagnose a disease or conditions in a non-serious situation or condition is a Category II and is considered to be of medium impact. Finally, SAMD of category I provides information to drive clinical management of a disease or conditions in a non-serious situation; this is considered a device of low risk.
SAMD and Classification According to EU MDR 2017/745
With the new MDR coming to life in the coming months, it is necessary to discuss about the new requirements introduced in this regulation in relation to the Software As Medical Devices. In fact, the key change introduced by the EU MDR for SAMD is related to the classification of medical software.
Specifically, going to review the classification rules associated to the EU MDR 2017/745, the rule 11 covers the classification of medical software:
Rule 11 Software intended to provide information which is used to take decisions with diagnosis or therapeutic purposes is classified as class IIa, except if such decisions have an impact that may cause: - death or an irreversible deterioration of a person’s state of health, in which case it is in class III; or - a serious deterioration of a person’s state of health or a surgical intervention, in which case it is classified as class IIb. Software intended to monitor physiological processes is classified as class IIa, except if it is intended for monitoring of vital physiological parameters, where the nature of variations of those parameters is such that it could result in immediate danger to the patient, in which case it is classified as class IIb. All other software is classified as class I.
The biggest change introduced by this classification rule is that software with a diagnostic or therapeutic purpose will fall into Class IIa (or higher). New rules for the classification of medical software will push some apps into a higher risk categorisation than they would have previously received. In the framework of the process for obtaining the CE marking, it means that many medical software that before were in self-certification (as Class I medical decides) now they are not any longer in this situation and CE marking can only be issued with the involvement of the notified body.
Software Development and Validation Package
4EasyReg has made available a toolkit focused on the documentation related to Software Development and Validation according to IEC 62304. We have been discussing in different occasion in the blog the documentation linked to IEC 62304, such as Software Development Plan and Software Architecture. We have however decide to collect the main documentation needed for activities related to Software Development and made it available in our 4EasyReg DocShop.
This toolkit contains different fully editable templates that can be used to document medical device related software development and validation activities.

The toolkit contains the following documentation:
- Software Development Plan Template
- Software Architecture Template
- Software Release Record Template
- Software Verification Protocol Template
- Software Verification Protocol Template
Do not hesitate to download it the toolkit!
Conclusions
We have been going through some important requirements associated to the software as medical device, with a specific focus on the European Medical Device Regulation. We described the methodology used for classification of the SAMD as proposed by the IMDF and evaluated the impact of medical device classification, in terms of class of risk, according to the EU MDR 2017/745.
Subscribe to 4EasyReg Newsletter
4EasyReg is an online platform dedicated to Regulatory matters within the medical device, information security and AI-Based business.
We offer a wide range of documentation kits to support your compliance efforts towards a wide range of standards and regulations, such as ISO 13485, EU MDR, ISO 27001, ISO 42001 and much more. . Specifically, in our webshop you will find:
- ISO 13485 Documentation / Compliance Kit
- EU MDR Documentation Kit
- MDSAP Documentation Kit
- ISO 27001 Documentation / Compliance Kit
- ISO 42001 Documentation / Compliance Kit
- FDA Cybersecurity Documentation
Within our sister platform QualityMedDev Academy, a wide range of online & self-paced training courses is available, such as for example:
- Complaint Handling and Vigilance Reporting
- Artificial Intelligence in Medical Device. Regulatory Requirements
- Unique Device Identification (UDI) Requirements according to EU MDR
- Clinical Evaluation Process According to EU MDR
- Medical Device SW Verification & Validation
- Risk Management for Medical Devices
- Usability Evaluation for Medical Devices
As one of the leading online platforms in the medical device sector, 4EasyReg offers extensive support for regulatory compliance. Our services cover a wide range of topics, from EU MDR & IVDR to ISO 13485, encompassing risk management, biocompatibility, usability, software verification and validation, and assistance in preparing technical documentation for MDR compliance.
Do not hesitate to subscribe to our Newsletter!
