One of the biggest changes introduced with the new IVDR 2017/746 is the classification for IVD products. In fact the classification substantially changes between IVDD and IVDR in order to be more aligned with the guideline published by the Global Harmonization Task Force (GHTF).
The methodology classification between standard medical device and in-vitro diagnostic devices is substantially different, because for the vast majority of the times, IVD device cannot directly harm the patient in the same way as a standard medical device potentially could.
This new classification, as mentioned before, is one of the key changes for the In-Vitro Diagnostic Regulations, along with other key changes such as the introduction of Periodic Safety Update Report and Post-Market Clinical Follow-up.
Overview of the IVDR Classification Rules
The IVDR 2017/746 establishes four risk classes D, C, B, and A, with D being the highest risk class and A the lowest. There are seven classification rules which are explained in the Annex VIII.
Given the complexity of the topics, the Medical Device Coordination Group published a specific document entitled Guidance on Classification Rules for in vitro Diagnostic Medical Devices under Regulation (EU) 2017/746 to further explained the strategy for the classification of IVD devices.
The seven rules for the classification of IVD devices can be summarised in the scheme below:
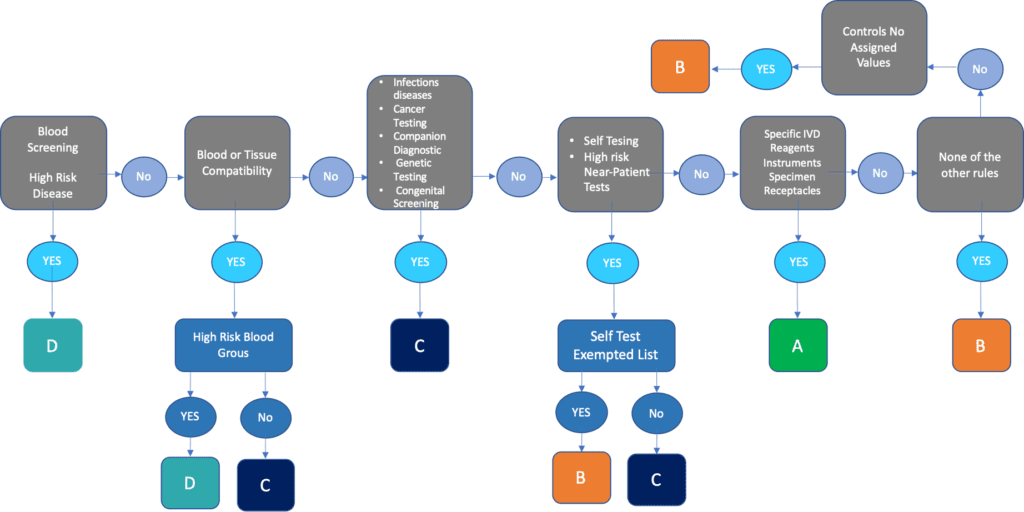
Class D In-Vitro diagnostic is related to life-threatening conditions and, in particular transmissible agents in blood and other biological materials intended to be re-inserted to administrated in the body.
Class C covers different of high-risk IVD devices which present a lesser risk to the wider population. These are devices where the failure of a diagnosis could be life-threatening, including testing for infectious diseases and cancer.
Class B is the default class that takes in all IVD devices that are not covered specifically in other classification rules. . It tends to cover devices that present lower risks to the patient and the population at large than IVD devices in Classes D and C. Class B also covers self-testing IVD devices for pregnancy and fertility testing as well as detection of cholesterol levels and detection of glucose, erythrocytes, leucocytes and bacteria in urine.
Class A covers broadly speaking laboratory devices, instruments and specimen receptacles.
Conformity Assessment
The conformity assessment is reported from Annexes IX to XI of the new EU IVDR 2017/746 and it is basically dependent from the class of risk of the IVD device.
The conformity assessment for Class D and class C devices is very similar and it can be based on two different options:
- 1st Option: the conformity assessment is based on Annex IX which includes Full Quality Management System Audit and random sampling of technical documentation
- 2nd Option: this conformity assessment is based on the combination of Type Examination (Annex X) and Production Quality Assurance (XI). EU type-examination is the procedure where a notified body ascertains that a device, including its technical documentation and relevant life cycle processes, fulfils the relevant provisions of this Regulation.
Class B IVD devices can be assessed through Annex IX, Chapters I and III (including an assessment of the technical documentation of at least one representative device per category of devices).
Finally, manufacturers of class A devices shall declare the conformity of their products by issuing the EU declaration of conformity referred to in Article 17 of the IVDR 2017/746.
Conclusions
In conclusions, we have been showing a summary of the classification rules and the related conformity assessment route for IVD device based on the In-Vitro Diagnostic Regulation 2017/746.
